首个获得定制化体内基因编辑治疗的新生儿
作者:jsba 发布日期:2025-05-19
近日,一例患儿创造了历史,他成为第一个接受定制化体内CRISPR基因编辑疗法治疗的患者。
患儿名为KJ,去年8月初出生时即被诊断为氨基甲酰磷酸合成酶1(CPS1)缺乏症,这是一种罕见的常染色体隐性遗传代谢疾病。CPS1基因突变导致患者无法正常代谢蛋白质分解产生的含氮废物(如氨),从而引发高血氨症,严重损害大脑功能。早发型的CPS1缺乏症病死率极高,病情进展迅速。
此类患者需依赖严格的低蛋白饮食、氮清除剂及可能的肝移植,但肝移植等待时间长、风险高,且无法根治。
而基因治疗扭转了这一命运。KJ现在已经10个月大,已能独坐、挥手、翻身,他即将首次回家与三个哥哥姐姐团聚。
这是全球首例针对个体患者特定基因突变定制的CRISPR疗法,从基因测序、疗法设计到临床审批仅耗时6个月,相关成果最近发表在了NEJM上。

明星班底提供定制基因编辑治疗
实际上,KJ出生不到48小时就已经出现了嗜睡、呼吸窘迫,血氨>1000 μmol/L(正常9-33 μmol/L)等症状。好在医生及时认出了该疾病。
母亲Nicole Muldoon坦言:“若非医生及时识别异常,他可能活不过第五天。”
标准处理显然治标不治本,患者的状况在与事件赛跑,在这种情况下,负责患儿的医生费城儿童医院(CHOP)代谢病专家Rebecca Ahrens-Nicklas几天后联系了基因治疗先驱Kiran Musunuru(Verve Therapeutics联合创始人)寻求帮助。
在这一背景下,一个跨学科,跨越多家技术公司的团队成立了。
此次研究由NIH直接资助,Acuitas Therapeutics提供LNP技术、Integrated DNA Technologies提供gRNA合成、Aldevron负责mRNA生产。
时间确实不等人。
患儿经连续肾脏替代治疗(CRRT)稳定后,依赖氮清除剂(苯丁酸甘油酯)、瓜氨酸补充及维持严格低蛋白饮食来稳定病情。
通过标准治疗,患儿在出生50-100天后经历短暂的稳定期,然而此后,血氨与谷氨酰胺水平再次升高,需进一步限制蛋白质摄入并增加药物剂量,这使得患儿最终在5月龄被列入肝移植名单。
留给团队的时间不多了。
治疗与研究历程
尽管KJ携带Q335X(父源)与E714X(母源)双突变,研究团队聚焦于其中更为关键的Q335X,修复Q335X突变后代谢获益更加明显。
由于无法直接获得患儿肝细胞,研究团队使用HuH-7细胞系构建携带CPS1 Q335X突变的体外模型,通过慢病毒载体将突变序列插入基因组,筛选最优碱基编辑器。
研究人员从多种腺嘌呤碱基编辑器(ABE)中选定NGC-ABE8e-V106W,其能够引导RNA(gRNA“kayjayguran”)靶向Q335X突变的第8位腺嘌呤,虽存在邻近腺嘌呤的“旁观者编辑”,但均为同义突变(同义突变不会改变蛋白质序列)。递送系统则采用的LNP-mRNA技术。
研究人员随后在小鼠和非人灵长类(食蟹猴)中进行了安全性及有效性验证。碱基编辑效率最高达实现了42%肝细胞修复。食蟹猴单剂量毒性试验显示,1.5 mg/kg RNA剂量的脂质纳米颗粒(LNP)仅引起短暂肝酶升高(ALT/AST),无其他毒性,支持人体低剂量(0.1 mg/kg)的安全性。
在随后的IND审查中,研究直接通过FDA的单患者扩大使用IND(研究性新药)申请获批,从提交到批准仅1周。
而伦理审查由CHOP机构审查委员会(IRB)通过替代程序加速完成,患儿父母签署书面知情同意。
具体注输方式采用了“低剂量启动”+“逐级递增”的模式。
在出生后第208天(也就是患儿被列入肝移植名单一个月后),患儿接受了首次注输,首次注输联合了免疫抑制剂(西罗莫司和他克莫司)来预防针对CPS1蛋白的免疫反应。治疗后蛋白质耐受性改善,但未能完全停用氮清除剂。
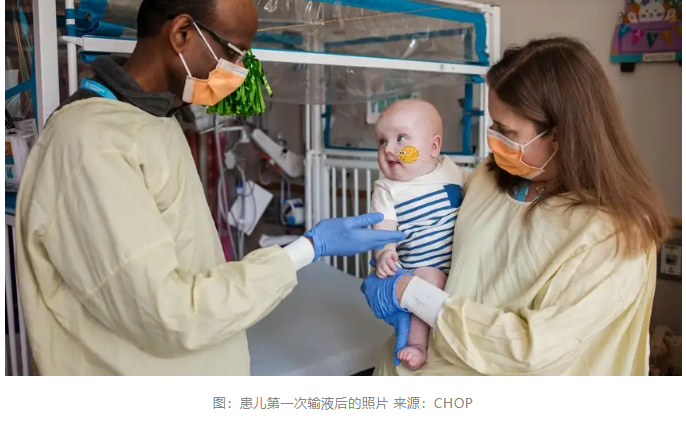
22天后的第二次注输,注输剂量提升,患儿血氨中位数从首次治疗前的23 μmol/L降至13 μmol/L,患儿尿乳清酸水平升至正常上限,这表明患儿CPS1功能已经得到部分恢复。患儿经历两次病毒感染仍保持数值稳定,无需中断蛋白质摄入。
第三次注输后,巩固疗效,最终使血氨稳定在近正常水平。
目前患儿的随访仅7周,还需长期监测编辑效率是否随肝细胞再生下降,以及是否需要补充剂量。另外患儿的母源突变是否会对患儿状况造成影响也值得关注。
总结
KJ案例不仅代表了体内基因编辑技术突破,更是监管创新与多学科协作的典范。从构建替代细胞模型、优化碱基编辑器到加速审批,团队在极短时间内实现“个体化疗法从设计到临床”的全流程验证。尽管长期安全性和可推广性还需要研究(此次个体案例花费超过100万美元),此案例为罕见病基因治疗提供了可复制的范式。( 来源:生物制药小编 )
参考来源:
doi: 10.1056/NEJMoa2504747